| [1] |
Berner R A. Early diagenesis: A theoretical approach[M]. New Jersey: Princeton University Press, 1980. |
| [2] |
Henrichs S M, 1992: Early diagenesis of organic matter in marine sediments progress and perplexity[J]. Marine Chemistry, 39, 119-149.
|
| [3] |
吴雪停,刘丽华,吴能友,等. 海洋沉积物中早期成岩作用地球化学研究进展[J]. 海洋地质前沿,2015,31(12):17-26. |
Wu Xueting, Liu Lihua, Wu Nengyou, et al. Geochemistry of early diagenesis in marine sediments: Research progress[J]. Marine Geology Frontiers, 2015, 31(12): 17-26. |
| [4] |
Canfield D E, Thamdrup B, 2009: Towards a consistent classification scheme for geochemical environments, or, why we wish the term ‘suboxic’ would go away[J]. Geobiology, 7, 385-392.
|
| [5] |
Froelich P N, Klinkhammer G P, Bender M L, 1979: Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: Suboxic diagenesis[J]. Geochimica et Cosmochimica Acta, 43, 1075-1090.
|
| [6] |
Hesse R, Schacht U, 2011: Early diagenesis of deep-sea sediments[J]. Developments in Sedimentology, 63, 557-713.
|
| [7] |
Aller R C. Sedimentary diagenesis, depositional environments, and benthic fluxes[M]//Holland H D, Turekian K K. Treatise on geochemistry.Oxford: Elsevier,2014: 293-334. |
| [8] |
Arndt S, Jørgensen B B, LaRowe D E, 2013: Quantifying the degradation of organic matter in marine sediments: A review and synthesis[J]. Earth-Science Reviews, 123, 53-86.
|
| [9] |
Warnken K W, Santschi P H, Roberts K A, 2008: The cycling and oxidation pathways of organic carbon in a shallow estuary along the Texas Gulf Coast[J]. Estuarine, Coastal and Shelf Science, 76, 69-84.
|
| [10] |
朱茂旭,史晓宁,杨桂朋,等. 海洋沉积物中有机质早期成岩矿化路径及其相对贡献[J]. 地球科学进展,2011,26(4):355-364. |
Zhu Maoxu, Shi Xiaoning, Yang Guipeng, et al. Relative contributions of various early diagenetic pathways to mineralization of organic matter in marine sediments: An overview[J]. Advances in Earth Science, 2011, 26(4): 355-364. |
| [11] |
Hensen C, Zabel M, Schulz H N. Benthic cycling of oxygen, nitrogen and phosphorus[M]//Schulz H D, Zabel M.Marine geochemistry. Berlin:Springer, 2006: 207-240. |
| [12] |
Middelburg J J, Soetaert K, Herman P M J, 1996: Denitrification in marine sediments: A model study[J]. Global Biogeochemical Cycles, 10, 661-673.
|
| [13] |
Lovley D R, Holmes D E, Nevin K P, 2004: Dissimilatory Fe(III) and Mn(IV) reduction[J]. Advances in Microbial Physiology, 49, 219-286.
|
| [14] |
Canfield D E, 1989: Reactive iron in marine sediments[J]. Geochimica et Cosmochimica Acta, 53, 619-632.
|
| [15] |
Zopfi J, Böttcher M E, Jørgensen B B, 2008: Biogeochemistry of sulfur and iron in Thioploca[J]. Geochimica et Cosmochimica Acta, 72, 827-843.
|
| [16] |
Canfield D E, Raiswell R, Bottrell S H, 1992: The reactivity of sedimentary iron minerals toward sulfide[J]. American Journal of Science, 292, 659-683.
|
| [17] |
Burdige D J, Nealson K H, 1986: Chemical and microbiological studies of sulfide‐mediated manganese reduction[J]. Geomicrobiology Journal, 4, 361-387.
|
| [18] |
Canfield D E, Thamdrup B, Hansen J W, 1993: The anaerobic degradation of organic matter in Danish coastal sediments: Iron reduction, manganese reduction, and sulfate reduction[J]. Geochimica et Cosmochimica Acta, 57, 3867-3883.
|
| [19] |
de Schamphelaire L, Rabaey K, Boon N, 2007: Minireview: The potential of enhanced manganese redox cycling for sediment oxidation[J]. Geomicrobiology Journal, 24, 547-558.
|
| [20] |
Thamdrup B. Bacterial manganese and iron reduction in aquatic sediments[M]//Schink B. Advances in microbial ecology.Boston:Springer,2000: 41-84. |
| [21] |
Thamdrup B, Glud R N, Hansen J W, 1994: Manganese oxidation and in situ manganese fluxes from a coastal sediment[J]. Geochimica et Cosmochimica Acta, 58, 2563-2570.
|
| [22] |
Aller R C, 1994: The sedimentary Mn cycle in Long Island Sound: Its role as intermediate oxidant and the influence of bioturbation, O2org[J]. Journal of Marine Research, 52, 259-295.
|
| [23] |
Canfield D E, Jørgensen B B, Fossing H, 1993: Pathways of organic carbon oxidation in three continental margin sediments[J]. Marine Geology, 113, 27-40.
|
| [24] |
Taylor K G, Macquaker J H S, 2011: Iron minerals in marine sediments record chemical environments[J]. Elements, 7, 113-118.
|
| [25] |
Zhao B, Yao P, Bianchi T S, 2018: The remineralization of sedimentary organic carbon in different sedimentary regimes of the Yellow and East China Seas[J]. Chemical Geology, 495, 104-117.
|
| [26] |
施春华,颜佳新,韩欣. 早期成岩作用过程中硫酸盐还原反应研究进展[J]. 广西地质,2001,14(1):21-26. |
Shi Chunhua, Yan Jiaxin, Han Xin. Development of sulfate reduction during early diagenesis[J]. Geology of Guangxi, 2001,14(1): 21-26. |
| [27] |
Canfield D E, 1991: Sulfate reduction in deep-sea sediments[J]. American Journal of Science, 291, 177-188.
|
| [28] |
Borowski W S, Paull C K, Ussler III W, 1996: Marine pore-water sulfate profiles indicate in situ methane flux from underlying gas hydrate[J]. Geology, 24, 655-658.
|
| [29] |
Aharon P, Fu B S, 2000: Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deep-water gulf of Mexico[J]. Geochimica et Cosmochimica Acta, 64, 233-246.
|
| [30] |
Canfield D E, 2001: Biogeochemistry of sulfur isotopes[J]. Reviews in Mineralogy and Geochemistry, 43, 607-636.
|
| [31] |
刘喜停,李安春,马志鑫,等. 沉积过程对自生黄铁矿硫同位素的约束[J]. 沉积学报,2020,38(1):124-137. |
Liu Xiting, Li Anchun, Ma Zhixin, et al. Constraint of sedimentary processes on the sulfur isotope of authigenic pyrite[J]. Acta Sedimentologica Sinica, 2020, 38(1): 124-137. |
| [32] |
Jørgensen B B, Beulig F, Egger M, 2019: Organoclastic sulfate reduction in the sulfate-methane transition of marine sediments[J]. Geochimica et Cosmochimica Acta, 254, 231-245.
|
| [33] |
Milucka J, Ferdelman T G, Polerecky L, 2012: Zero-valent sulphur is a key intermediate in marine methane oxidation[J]. Nature, 491, 541-546.
|
| [34] |
Zigah P K, Oswald K, Brand A, 2015: Methane oxidation pathways and associated methanotrophic communities in the water column of a tropical lake[J]. Limnology and Oceanography, 60, 553-572.
|
| [35] |
Haroon M F, Hu S H, Shi Y, 2013: Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage[J]. Nature, 500, 567-570.
|
| [36] |
Ettwig K F, Butler M K, Le Paslier D, 2010: Nitrite-driven anaerobic methane oxidation by oxygenic bacteria[J]. Nature, 464, 543-548.
|
| [37] |
Rooze J, Egger M, Tsandev I, 2016: Iron-dependent anaerobic oxidation of methane in coastal surface sediments: Potential controls and impact[J]. Limnology and Oceanography, 61, S267-S282.
|
| [38] |
Liu J R, Izon G, Wang J S, 2018: Vivianite formation in methane-rich deep-sea sediments from the South China Sea[J]. Biogeosciences, 15, 6329-6348.
|
| [39] |
Egger M, Kraal P, Jilbert T, 2016: Anaerobic oxidation of methane alters sediment records of sulfur, iron and phosphorus in the Black Sea[J]. Biogeosciences, 13, 5333-5355.
|
| [40] |
Jing H M, Wang R N, Jiang Q Y, 2020: Anaerobic methane oxidation coupled to denitrification is an important potential methane sink in deep-sea cold seeps[J]. Science of the Total Environment, 748, 142459-.
|
| [41] |
Ettwig K F, Zhu B L, Speth D, 2016: Archaea catalyze iron-dependent anaerobic oxidation of methane[J]. Proceedings of the National Academy of Sciences of the United States of America, 113, 12792-12796.
|
| [42] |
Devol A H, Anderson J J, Kuivila K, 1984: A model for coupled sulfate reduction and methane oxidation in the sediments of Saanich Inlet[J]. Geochimica et Cosmochimica Acta, 48, 993-1004.
|
| [43] |
Kuivila K M, Murray J W, Devol A H, 1989: Methane production, sulfate reduction and competition for substrates in the sediments of Lake Washington[J]. Geochimica et Cosmochimica Acta, 53, 409-416.
|
| [44] |
Reeburgh W S, 2007: Oceanic methane biogeochemistry[J]. Chemical Reviews, 107, 486-513.
|
| [45] |
Rust G W, 1935: Colloidal primary copper ores at Cornwall mines, southeastern Missouri[J]. The Journal of Geology, 43, 398-426.
|
| [46] |
Rickard D, 2019: Sedimentary pyrite framboid size-frequency distributions: A meta-analysis[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 522, 62-75.
|
| [47] |
Oenema O, 1990: Pyrite accumulation in salt marshes in the eastern Scheldt, southwest Netherlands[J]. Biogeochemistry, 9, 75-98.
|
| [48] |
Allen K D, Hahn G A, 1994: Geology of the sunbeam and grouse creek gold-silver deposits, Yankee Fork mining district, Eocene Challis volcanic field, Idaho; a volcanic dome- and volcaniclastic-hosted epithermal system[J]. Economic Geology, 89, 1964-1982.
|
| [49] |
Berner R A, 1970: Sedimentary pyrite formation[J]. American Journal of Science, 268, 1-23.
|
| [50] |
Wilkin R T, Barnes H L, 1997: Formation processes of framboidal pyrite[J]. Geochimica et Cosmochimica Acta, 61, 323-339.
|
| [51] |
Lan Y, Butler E C, 2014: Monitoring the transformation of mackinawite to greigite and pyrite on polymer supports[J]. Applied Geochemistry, 50, 1-6.
|
| [52] |
Sugawara H, Sakakibara M, Belton D, 2013: Formation process of pyrite polyframboid based on the heavy-metal analysis by micro-PIXE[J]. Environmental Earth Sciences, 69, 811-819.
|
| [53] |
Huang H W, Gong S G, Li N, 2019: Formation of authigenic low-magnesium calcite from sites SS296 and GC53 of the gulf of Mexico[J]. Minerals, 9, 251-.
|
| [54] |
Tong H P, Feng D, Peckmann J, 2019: Environments favoring dolomite formation at cold seeps: A case study from the gulf of Mexico[J]. Chemical Geology, 518, 9-18.
|
| [55] |
冯东,陈多福,漆亮,等. 墨西哥湾Alaminos Canyon冷泉碳酸盐岩地质地球化学特征[J]. 科学通报,2008,53(8):966-974. |
Feng Dong, Chen Duofu, Qi Liang, et al. Geological and geochemical characteristics of Alaminos Canyon cold spring carbonates in the gulf of Mexico[J]. Scientific Bulletin, 2008, 53(8): 966-974. |
| [56] |
赵洁,王家生,岑越,等. 南海东北部GMGS2-16站位自生矿物特征及对水合物藏演化的指示意义[J]. 海洋地质与第四纪地质,2018,38(5):144-155. |
Zhao Jie, Wang Jiasheng, Cen Yue, et al. Authigenic minerals at site GMGS2-16 of northeastern South China Sea and its implications for gas hydrate evolution[J]. Marine Geology & Quaternary Geology, 2018, 38(5): 144-155. |
| [57] |
Han X Q, Suess E, Huang Y Y, 2008: Jiulong methane reef: Microbial mediation of seep carbonates in the South China Sea[J]. Marine Geology, 249, 243-256.
|
| [58] |
Magalhães V H, Pinheiro L M, Ivanov M K, 2012: Formation processes of methane-derived authigenic carbonates from the gulf of Cadiz[J]. Sedimentary Geology, 243-244, 155-168.
|
| [59] |
Naehr T H, Eichhubl P, Orphan V J, 2007: Authigenic carbonate formation at hydrocarbon seeps in continental margin sediments: A comparative study[J]. Deepsea Research Part II: Topical Studies in Oceanography, 54, 1268-1291.
|
| [60] |
Baker J C, Kassan J, Hamilton P J, 1996: Early diagenetic siderite as an indicator of depositional environment in the Triassic Rewan Group, southern Bowen Basin, eastern Australia[J]. Sedimentology, 43, 77-88.
|
| [61] |
陈惠昌,赖勇,卢海龙,等. 南海神狐天然气水合物系统沉积物中自生黄铁矿的特征研究[J]. 海洋学报,2018,40(7):116-133. |
Chen Huichang, Lai Yong, Lu Hailong, et al. Study on authigenic pyrite in sediments of gas hydrate geo-system in the Shenhu area, South China Sea[J]. Haiyang Xuebao, 2018, 40(7): 116-133. |
| [62] |
Taylor K G, Curtis C D, 1995: Stability and facies association of early diagenetic mineral assemblages; an example from a Jurassic ironstone-mudstone succession, U.K[J]. Journal of Sedimentary Research, 65, 358-368.
|
| [63] |
Mozley P S, Carothers W W, 1992: Elemental and isotopic composition of siderite in the Kuparuk Formation, Alaska; effect of microbial activity and water sediment interaction on early pore-water chemistry[J]. Journal of Sedimentary Research, 62, 681-692.
|
| [64] |
Whiticar M J, 1999: Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane[J]. Chemical Geology, 161, 291-314.
|
| [65] |
Feng D, Qiu J W, Hu Y, 2018: Cold seep systems in the South China Sea: An overview[J]. Journal of Asian Earth Sciences, 168, 3-16.
|
| [66] |
Campbell K A, 2006: Hydrocarbon seep and hydrothermal vent paleoenvironments and paleontology: Past developments and future research directions[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 232, 362-407.
|
| [67] |
Greinert J, Bohrmann G, Suess E, 2013: Gas hydrate‐associated carbonates and methane‐venting at hydrate ridge: Classification, distribution, and origin of authigenic lithologies[J]. Geophysical MonographSeries, 124, 99-113.
|
| [68] |
Peckmann J, Thiel V, 2004: Carbon cycling at ancient methane-seeps[J]. Chemical Geology, 205, 443-467.
|
| [69] |
Mansour A S, Sassen R, 2011: Mineralogical and stable isotopic characterization of authigenic carbonate from a hydrocarbon seep site, gulf of Mexico slope: Possible relation to crude oil degradation[J]. Marine Geology, 281, 59-69.
|
| [70] |
Mozley P S, Wersin P, 1992: Isotopic composition of siderite as an indicator of depositional environment[J]. Geology, 20, 817-820.
|
| [71] |
Mozley P S, 1989: Relation between depositional environment and the elemental composition of early diagenetic siderite[J]. Geology, 17, 704-706.
|
| [72] |
张凌,陈繁荣,杨永强,等. 珠江口外近海沉积有机质化学及稳定同位素组成的早期成岩改变[J]. 海洋学报,2008,30(5):43-51. |
Zhang Ling, Chen Fanrong, Yang Yongqiang, et al. Chemical and isotopic alteration of organic matter during early diagenesis: Evidence from the coastal area offshore the Zhujiang Estuary in China[J]. Acta Oceanologica Sinica, 2008, 30(5): 43-51. |
| [73] |
Lehmann M F, Bernasconi S M, Barbieri A, 2002: Preservation of organic matter and alteration of its carbon and nitrogen isotope composition during simulated and in situ early sedimentary diagenesis[J]. Geochimica et Cosmochimica Acta, 66, 3573-3584.
|
| [74] |
DeNiro M J, Epstein S, 1977: Mechanism of carbon isotope fractionation associated with lipid synthesis[J]. Science, 197, 261-263.
|
| [75] |
Macko S A, Estep M L F, 1984: Microbial alteration of stable nitrogen and carbon isotopic compositions of organic matter[J]. Organic Geochemistry, 6, 787-790.
|
| [76] |
Prahl F G, de Lange G J, Scholten S, 1997: A case of post-depositional aerobic degradation of terrestrial organic matter in turbidite deposits from the Madeira Abyssal Plain[J]. Organic Geochemistry, 27, 141-152.
|
| [77] |
Sachs J P, Repeta D J, 1999: Oligotrophy and nitrogen fixation during eastern Mediterranean Sapropel events[J]. Science, 286, 2485-2488.
|
| [78] |
Freudenthal T, Wagner T, Wenzhofer F, 2001: Early diagenesis of organic matter from sediments of the eastern subtropical Atlantic: Evidence from stable nitrogen and carbon isotopes[J]. Geochimica et Cosmochimica Acta, 65, 1795-1808.
|
| [79] |
Altabet M A, Pilskaln C, Thunell R, 1999: The nitrogen isotope biogeochemistry of sinking particles from the margin of the eastern North Pacific[J]. Deep Sea Research Part I: Oceanographic Research Papers, 46, 655-679.
|
| [80] |
Goldhaber M B, 2003: Sulfur-rich sediments[J]. Treatise on Geochemistry, 7, 257-288.
|
| [81] |
Rickard D, Morse J W, 2005: Acid volatile sulfide (AVS)[J]. Marine Chemistry, 97, 141-197.
|
| [82] |
Böttcher M E, Lepland A, 2000: Biogeochemistry of sulfur in a sediment core from the west-central Baltic Sea: Evidence from stable isotopes and pyrite textures[J]. Journal of Marine Systems, 25, 299-312.
|
| [83] |
Wilkin R T, Arthur M A, 2001: Variations in pyrite texture, sulfur isotope composition, and iron systematics in the Black Sea: Evidence for Late Pleistocene to Holocene excursions of the O22[J]. Geochimica et Cosmochimica Acta, 65, 1399-1416.
|
| [84] |
Lin Z Y, Sun X M, Peckmann J, 2016: How sulfate-driven anaerobic oxidation of methane affects the sulfur isotopic composition of pyrite: A SIMS study from the South China Sea[J]. Chemical Geology, 440, 26-41.
|
| [85] |
Li N, Feng D, Chen L Y, 2016: Using sediment geochemistry to infer temporal variation of methane flux at a cold seep in the South China Sea[J]. Marine and Petroleum Geology, 77, 835-845.
|
| [86] |
Lin Z Y, Sun X M, Lu Y, 2016: Stable isotope patterns of coexisting pyrite and gypsum indicating variable methane flow at a seep site of the Shenhu area, South China Sea[J]. Journal of Asian Earth Sciences, 123, 213-223.
|
| [87] |
Hu Y, Chen L Y, Feng D, 2017: Geochemical record of methane seepage in authigenic carbonates and surrounding host sediments: A case study from the South China Sea[J]. Journal of Asian Earth Sciences, 138, 51-61.
|
| [88] |
Lin Q, Wang J S, Taladay K, 2016: Coupled pyrite concentration and sulfur isotopic insight into the paleo sulfate-methane transition zone (SMTZ) in the northern South China Sea[J]. Journal of Asian Earth Sciences, 115, 547-556.
|
| [89] |
Canfield D E, Thamdrup B, 1994: The production of 34[J]. Science, 266, 1973-1975.
|
| [90] |
Wortmann U G, Bernasconi S M, Böttcher M E, 2001: Hypersulfidic deep biosphere indicates extreme sulfur isotope fractionation during single-step microbial sulfate reduction[J]. Geology, 29, 647-650.
|
| [91] |
Sim M S, Bosak T, Ono S, 2011: Large sulfur isotope fractionation does not require disproportionation[J]. Science, 333, 74-77.
|
| [92] |
Canfield D E, Farquhar J, Zerkle A L, 2010: High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog[J]. Geology, 38, 415-418.
|
| [93] |
Deusner C, Holler T, Arnold G L, 2014: Sulfur and oxygen isotope fractionation during sulfate reduction coupled to anaerobic oxidation of methane is dependent on methane concentration[J]. Earth and Planetary Science Letters, 399, 61-73.
|
| [94] |
Sivan O, Antler G, Turchyn A V, 2014: Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps[J]. Proceedings of the National Academy of Sciences of the United States of America, 111, E4139-E4147.
|
| [95] |
Borowski W S, Rodriguez N M, Paull C K, 2013: Are 34[J]. Marine and Petroleum Geology, 43, 381-395.
|
| [96] |
Fritz P, Basharmal G M, Drimmie R J, 1989: Oxygen isotope exchange between sulphate and water during bacterial reduction of sulphate[J]. Chemical Geology: Isotope Geoscience Section, 79, 99-105.
|
| [97] |
Brunner B, Bernasconi S M, Kleikemper J, 2005: A model for oxygen and sulfur isotope fractionation in sulfate during bacterial sulfate reduction processes[J]. Geochimica et Cosmochimica Acta, 69, 4773-4785.
|
| [98] |
Wortmann U G, Chernyavsky B, Bernasconi S M, 2007: Oxygen isotope biogeochemistry of pore water sulfate in the deep biosphere: Dominance of isotope exchange reactions with ambient water during microbial sulfate reduction (ODP Site 1130)[J]. Geochimica et Cosmochimica Acta, 71, 4221-4232.
|
| [99] |
Wankel S D, Bradley A S, Eldridge D L, 2014: Determination and application of the equilibrium oxygen isotope effect between water and sulfite[J]. Geochimica et Cosmochimica Acta, 125, 694-711.
|
| [100] |
Müller I A, Brunner B, Breuer C, 2013: The oxygen isotope equilibrium fractionation between sulfite species and water[J]. Geochimica et Cosmochimica Acta, 120, 562-581.
|
| [101] |
van Cappellen P, Ingall E D, 1994: Benthic phosphorus regeneration, net primary production, and ocean anoxia: A model of the coupled marine biogeochemical cycles of carbon and phosphorus[J]. Paleoceanography, 9, 677-692.
|
| [102] |
刘喜停,颜佳新. 铁元素对海相沉积物早期成岩作用的影响[J]. 地球科学进展,2011,26(5):482-492. |
Liu Xiting, Yan Jiaxin. Advances in the role of iron in marine sediments during early diagenesis[J]. Advances in Earth Science, 2011, 26(5): 482-492. |
| [103] |
Beard B L, Johnson C M, Cox L, 1999: Iron isotope biosignatures[J]. Science, 285, 1889-1892.
|
| [104] |
Johnson C M, Roden E E, Welch S A, 2005: Experimental constraints on Fe isotope fractionation during magnetite and Fe carbonate formation coupled to dissimilatory hydrous ferric oxide reduction[J]. Geochimica et Cosmochimica Acta, 69, 963-993.
|
| [105] |
Calvert S E, Pedersen T F, 1993: Geochemistry of recent oxic and anoxic marine sediments: Implications for the geological record[J]. Marine Geology, 113, 67-88.
|
| [106] |
Calvert S E, Pedersen T F, 1996: Sedimentary geochemistry of manganese; implications for the environment of formation of manganiferous black shales[J]. Economic Geology, 91, 36-47.
|
| [107] |
Rajendran A, Kumar M D, Bakker J F, 1992: Control of manganese and iron in Skagerrak sediments (northeastern North Sea)[J]. Chemical Geology, 98, 111-129.
|
| [108] |
Morford J L, Russell A D, Emerson S, 2001: Trace metal evidence for changes in the redox environment associated with the transition from terrigenous clay to diatomaceous sediment, Saanich Inlet, BC[J]. Marine Geology, 174, 355-369.
|
| [109] |
Caplan M L, Bustin R M, 2001: Palaeoenvironmental and palaeoceanographic controls on black, laminated mudrock deposition: Example from Devonian-Carboniferous strata, Alberta, Canada[J]. Sedimentary Geology, 145, 45-72.
|
| [110] |
Cruse A M, Lyons T W, 2004: Trace metal records of regional paleoenvironmental variability in Pennsylvanian (Upper Carboniferous) black shales[J]. Chemical Geology, 206, 319-345.
|
| [111] |
Tribovillard N, Algeo T J, Lyons T, 2006: Trace metals as paleoredox and paleoproductivity proxies: An update[J]. Chemical Geology, 232, 12-32.
|
| [112] |
鲍根德. 铁、锰在早期成岩过程中分离及其生物地球化学机制[J]. 中国科学:B辑,1989(1):93-102. |
Bao Gende. Separation of iron and manganese in the early diagenetic processes and its mechanism of biogeochemistry[J]. Science in China(Seri.B), 1989(1): 93-102. |
| [113] |
Madison A S, Tebo B M, Mucci A, 2013: Abundant porewater Mn(III) is a major component of the sedimentary redox system[J]. Science, 341, 875-878.
|
| [114] |
Tyson R V, Pearson T H, 1991: Modern and ancient continental shelf anoxia: An overview[J]. Geological Society, London, Special Publications, 58, 1-24.
|
| [115] |
颜佳新,张海清. 古氧相:一个新的沉积学研究领域[J]. 地质科技情报,1996,15(3):8-14. |
Yan Jiaxin, Zhang Haiqing. Paleo-oxygenation facies: A new research field in sedimentology[J]. Geological Science and Technology Information, 1996, 15(3): 8-14. |
| [116] |
汤冬杰,史晓颖,赵相宽,等. Mo-U共变作为古沉积环境氧化还原条件分析的重要指标:进展、问题与展望[J]. 现代地质,2015,29(1):1-13. |
Tang Dongjie, Shi Xiaoying, Zhao Xiangkuan, et al. Mo-U covariation as an important proxy for sedimentary environment redox conditions-progress,problems and prospects[J]. Geoscience, 2015,29(1): 1-13. |
| [117] |
Lyons T W, Severmann S, 2006: A critical look at iron paleoredox proxies: New insights from modern euxinic marine basins[J]. Geochimica et Cosmochimica Acta, 70, 5698-5722.
|
| [118] |
Poulton S W, Canfield D E, 2011: Ferruginous conditions: A dominant feature of the ocean through Earth's history[J]. Elements, 7, 107-112.
|
| [119] |
Raiswell R, Hardisty D S, Lyons T W, 2018: The iron paleoredox proxies: A guide to the pitfalls, problems and proper practice[J]. American Journal of Science, 318, 491-526.
|
| [120] |
Raiswell R, Berner R A, 1985: Pyrite formation in euxinic and semi-euxinic sediments[J]. American Journal of Science, 285, 710-724.
|
| [121] |
Raiswell R, Buckley F, Berner R A, 1988: Degree of pyritization of iron as a paleoenvironmental indicator of bottom-water oxygenation[J]. Journal of Sedimentary Research, 58, 812-819.
|
| [122] |
Raiswell R, Canfield D E, 1998: Sources of iron for pyrite formation in marine sediments[J]. American Journal of Science, 298, 219-245.
|
| [123] |
Li C, Love G D, Lyons T W, 2010: A stratified redox model for the Ediacaran Ocean[J]. Science, 328, 80-83.
|
| [124] |
Poulton S W, Raiswell R, 2002: The low-temperature geochemical cycle of iron: From continental fluxes to marine sediment deposition[J]. American Journal of Science, 302, 774-805.
|
| [125] |
Poulton S W, Fralick P W, Canfield D E, 2010: Spatial variability in oceanic redox structure 1.8 billion years ago[J]. Nature Geoscience, 3, 486-490.
|
| [126] |
Redfield A C, 1958: The biological control of chemical factors in the environment[J]. American Scientist, 46, 205-221.
|
| [127] |
Algeo T J, Ingall E, 2007: Sedimentary Corg2[J]. Palaeogeography, Palaeoclimatology, Palaeoecology, 256, 130-155.
|
| [128] |
Steenbergh A K, Bodelier P L E, Hoogveld H L, 2011: Phosphatases relieve carbon limitation of microbial activity in Baltic Sea sediments along a redox-gradient[J]. Limnology and Oceanography, 56, 2018-2026.
|
| [129] |
Kraal P, Slomp C P, Reed D C, 2012: Sedimentary phosphorus and iron cycling in and below the oxygen minimum zone of the northern Arabian Sea[J]. Biogeosciences, 9, 2603-2624.
|
| [130] |
Algeo T J, Li C, 2020: Redox classification and calibration of redox thresholds in sedimentary systems[J]. Geochimica et Cosmochimica Acta, 287, 8-26.
|
| [131] |
Morford J L, Emerson S R, Breckel E J, 2005: Diagenesis of oxyanions (V, U, Re, and Mo) in pore waters and sediments from a continental margin[J]. Geochimica et Cosmochimica Acta, 69, 5021-5032.
|
| [132] |
Jones B, Manning D A C, 1994: Comparison of geochemical indices used for the interpretation of palaeoredox conditions in ancient mudstones[J]. Chemical Geology, 111, 111-129.
|
| [133] |
Algeo T J, Tribovillard N, 2009: Environmental analysis of paleoceanographic systems based on molybdenum-uranium covariation[J]. Chemical Geology, 268, 211-225.
|
| [134] |
Little S H, Vance D, Lyons T W, 2015: Controls on trace metal authigenic enrichment in reducing sediments: Insights from modern oxygen-deficient settings[J]. American Journal of Science, 315, 77-119.
|
| [135] |
Sweere T, van den Boorn S, Dickson A J, 2016: Definition of new trace-metal proxies for the controls on organic matter enrichment in marine sediments based on Mn, Co, Mo and Cd concentrations[J]. Chemical Geology, 441, 235-245.
|
| [136] |
Taylor S R, McLennan S M, 1985: The continental crust: Its composition and evolution[J]. The Journal of Geology, 94, 57-72.
|
| [137] |
McLennan S M, 2001: Relationships between the trace element composition of sedimentary rocks and upper continental crust[J]. Geochemistry, Geophysics, Geosystems, 2, 2000GC000109-.
|
| [138] |
张明亮,郭伟,沈俊,等. 古海洋氧化还原地球化学指标研究新进展[J]. 地质科技情报,2017,36(4):95-106. |
Zhang Mingliang, Guo Wei, Shen Jun, et al. New progress on geochemical indicators of ancient oceanic redox condition[J]. Geological Science and Technology Information, 2017, 36(4): 95-106. |
| [139] |
Noordmann J, Weyer S, Montoya-Pino C, 2015: Uranium and molybdenum isotope systematics in modern euxinic basins: Case studies from the central Baltic Sea and the Kyllaren fjord (Norway)[J]. Chemical Geology, 396, 182-195.
|
| [140] |
Barling J, Arnold G L, Anbar A D, 2001: Natural mass-dependent variations in the isotopic composition of molybdenum[J]. Earth and Planetary Science Letters, 193, 447-457.
|
| [141] |
Siebert C, Nägler T F, von Blanckenburg F, 2003: Molybdenum isotope records as a potential new proxy for paleoceanography[J]. Earth and Planetary Science Letters, 211, 159-171.
|
| [142] |
Poulson R L, Siebert C, McManus J, 2006: Authigenic molybdenum isotope signatures in marine sediments[J]. Geology, 34, 617-620.
|
| [143] |
Montoya-Pino C, Weyer S, Anbar A D, 2010: Global enhancement of ocean anoxia during Oceanic Anoxic Event 2: A quantitative approach using U isotopes[J]. Geology, 38, 315-318.
|
| [144] |
Romaniello S J, Herrmann A D, Anbar A D, 2013: Uranium concentrations and 238235[J]. Chemical Geology, 362, 305-316.
|
| [145] |
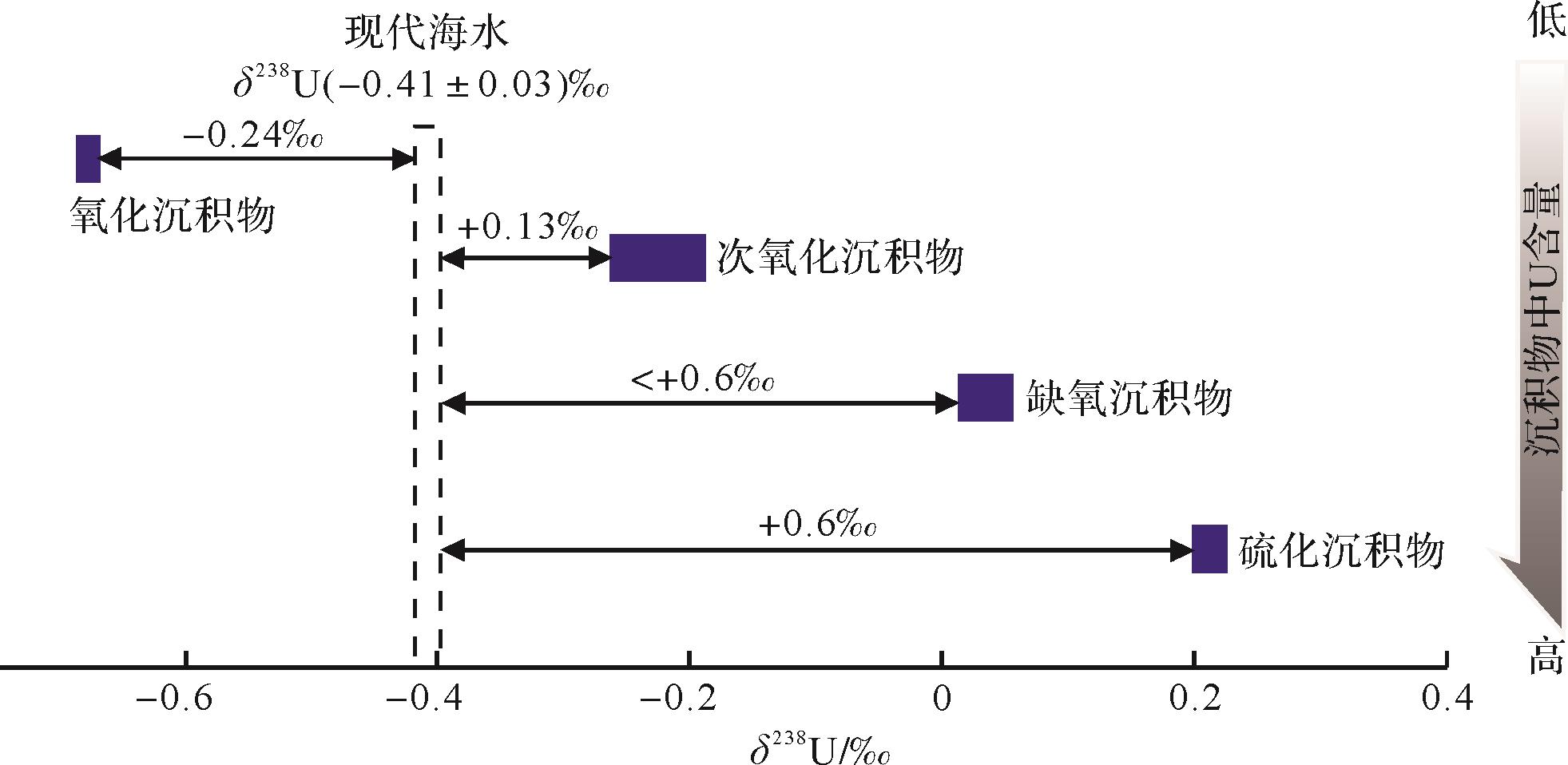
Goto K T, Anbar A D, Gordon G W, 2014: Uranium isotope systematics of ferromanganese crusts in the Pacific Ocean: Implications for the marine 238235[J]. Geochimica et Cosmochimica Acta, 146, 43-58.
|














 DownLoad:
DownLoad: